
Por RAQUEL LUIZA,
O termo hemostasia se refere ao conjunto de eventos fisiológicos que são acionados por uma lesão vascular. Juntamente com o fenômeno inflamatório, constitui um mecanismo de defesa do organismo que tende a reduzir e interromper a perda sanguínea, através da atividade coordenada de fatores vasculares, plaquetários e plasmáticos, cuja regulação e equilíbrio previnem déficit (hemorragia) ou acúmulo excessivo (trombose) de plaquetas e fibrina.
A coagulação sanguínea, um dos mais proeminentes e maiores aspectos desse processo, é realizada por várias pró-enzimas que circulam inativamente na corrente sanguínea e que, por meio de uma cascata de reações proteolíticas específicas, formam o coágulo de fibrina (via intrínseca e extrínseca).
A sequência de eventos após uma lesão vascular inclui: espasmo vascular (vasoconstrição), formação de tampão plaquetário (adesão e agregação plaquetária), formação de coágulo sanguíneo (cascata de coagulação) e eventual proliferação de tecido fibroso no interior do referido coágulo, a fim de definitivamente feche a solução de continuidade da parede vascular.
A hemostasia começa quando o fator de Von Willebrand adere ao colágeno exposto na ferida da parede vascular. O referido fator é uma molécula de plasma circulante, que se encontra tanto no interior da célula endotelial que o produz, quanto nas plaquetas. Tem a particularidade de cada uma das subunidades que o compõem estar associada à molécula do fator de coagulação VII, bem como a propriedade de aderir, por um lado, ao colágeno do subendotélio e, por outro, aos receptores que existem na membrana plaquetária (glicoproteínas Ib). As plaquetas aderidas ao colágeno do subendotélio mudam de forma e liberam seu conteúdo, principalmente ADP (difosfato de adenosina) e tromboxano A2, criando uma atmosfera de substâncias pró-agregantes que têm a capacidade de agregar maior quantidade de plaquetas em relação às já aderidas. Essas e outras substâncias liberadas pelas plaquetas aderidas alteram ainda mais sua forma e expõem outros tipos de receptores (glicoproteínas IIb – IIIa), que fazem as plaquetas aderirem umas às outras.
Essa adesão é chamada de agregação plaquetária e é realizada por pontes de fibronogênio. Ai, a molécula de fibrogênio se liga, por um lado, às glicoproteínas IIb-IIIa de uma plaqueta e, por outro lado, às glicoportinas IIb-IIIa de outra plaqueta. Isso produz um tampão de plaquetas nas plaquetas inicialmente aderidas, em cuja superfície rugosa será construída a próxima fase da hemostasia, ou seja, a formação do coágulo de fibrina.
FATORES DE PLAQUETAS
- Proteínas contráteis: Actina, Miosina, Trombostenina.
- Complexos enzimáticos (retículo endoplasmático e aparelho de Golgi)
- Depósitos de cálcio (restos de retículo endoplasmático)
- ATP e ADP (resíduos mitocondriais).
- Prostaglandinas, tromboxano A2.
- Fator de perseguição de fibrina.
- PGDF
- Glicoproteínas e fosfolipídios da membrana celular (adesão e ativação de fatores de coagulação).
MECANISMO DE ATIVAÇÃO DE PLAQUETAS E LIBERAÇÃO DE GRÂNULOS
A ativação das plaquetas começa com a união de um agonista à superfície plaquetária e a ativação das fosfolipases C e A2 pelas proteínas G. A partir deste ponto, uma cascata de reações bioquímicas leva à liberação de cálcio tanto do fluido extracelular como do reticuloendoplasmático depósitos, que atua como um mensageiro promovendo sinais elétricos que iniciam a sequência de ações tromocíticas.
As plaquetas circulantes entram em contato com as moléculas adesivas, especialmente o colágeno tipo IV. Isso é possível graças a uma proteína solúvel chamada fator de von Willebrand, que serve como ponte entre esses componentes e receptores específicos da membrana plaquetária (glicoproteína Ib). A plaqueta então libera mediadores químicos presentes em seus grânulos alfa, incluindo beta-microglobulina, fator plaquetário 4, trombospondina, fator de von willwbrand, fibrinogênio e PDGF (fator de crescimento derivado de plaquetas – fator de crescimento derivado de plaquetas). Ao mesmo tempo, ADP (adenosina disdofato), ATP (adenosina trifosfato), epinefrina e serotonina são liberados dos grânulos alfa, enquanto o tromboxano A2 é ativamente sintetizado pela enzima ciclooxigenase. Essas substâncias atuam em receptores de membrana especializados, recrutando um maior número de plaquetas. Ao mesmo tempo, outro receptor é exposto na superfície plaquetária, a glicoproteira IIb / IIIa, que intervém nos fenômenos de agregação criando pontes entre os elementos celulares adjacentes por meio do fibrogênio e do fator de von Willebrand.
Além disso, as plaquetas fornecem uma superfície rica em fosfolipídios, necessária à realização de várias etapas da cascata de coagulação e formação da trombina, proteína responsável pela transformação do fibrinogênio em fibrina.
A trombina, por sua vez, é o principal ativador das plaquetas e também estimula a expressão da glicoproteína IIb / IIIa.
Dessa forma, mostra-se como a coagulação e a agregação plaquetária são fenômenos interdependentes. Para controlar a adesão e o agravamento plaquetário, o endotélio intacto produz prostaciclina e óxido nítrico. Além disso, os glicosaminoglicanos presentes na membrana da célula endotelial têm carga negativa e repelem plaquetas que também possuem cargas negativas. Dessa forma, esses fenômenos limitam-se exclusivamente ao local da lesão.
FORMAÇÃO DO COAGÚLO DE FIBRINA – CASCATA DE COAGULAÇÃO
A contração vascular e a formação do tampão plaquetário são seguidos pela formação do coágulo sanguíneo, cujo aparecimento ocorre 15-20 segundos após a lesão se ela foi intensa, ou em 1-2 minutos, se foi leve. Dentre os fenômenos físicos desse processo, destacam-se a aglutinação de plaquetas, a formação do coágulo de fibrina, o surgimento da fibrina e a retração do coágulo, que tende a fechar ainda mais o vaso lesado. Sua evolução subsequente pode seguir vias, seja a invasão por fibroblastos e por sua completa organização pelo tecido conjuntivo em 1-2 semanas (mais freqüente em pequenas lesões), ou sua dissolução quando, por coagulação adicional de sangue extravasado aos tecidos em lesões maiores , são ativadas substâncias especiais que atuam como catalisadores enzimáticos.
O fator VIII está envolvido na ativação do fator X na sequência que leva à formação do coágulo de fibrina, circula associado ao fator de von Willebran, que atua como estabilizador, é produzido sob o controle de genes autossômicos e é encontrado no plasma, megacariócitos, plaquetas e células endoteliais. A hemofilia A é causada tanto pela diminuição dos níveis de fator VIII quanto pela presença de moléculas anormais. Múltiplas alterações no nível genético (deleções, replicações, inserções e mutações pontuais, inversões, etc.), distribuídas por todo o gene do fator VIII, foram reconhecidas como causadoras desse defeito.
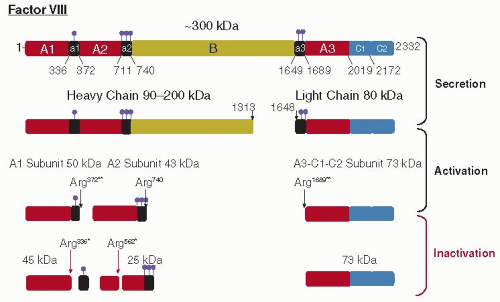
O fator VIII é uma glicoproteína sintetizada em hepacitos e células endoteliais, inicialmente como uma proteína precursora de cadeia simples de 2.351 aminoácidos e peso molecular de 265.000 Da. A molécula original intacta compreende três tipos de domínios, conforme mostrado neste esquema:
– Domínio A: três segmentos A (A1, A2, A3), cada um com aproximadamente 330 aminoácidos.
-Domínio B: um único segmento central que consiste em 980 aminoácidos.
-Domínio C: dois segmentos C (C1, C2) de 150 aminoácidos cada.
No plasma, a molécula do fator VIII é clivada para produzir dois peptídeos, uma cadeia pesada do terminal animo (domínios A1, A2, B) e uma cadeia leve do terminal carboxi (A3, C1, C2). A proteína circula como um dímero, com íons de cálcio ligando as duas cadeias pesadas e é clivada, liberando o domínio B, que não é necessário para a atividade de coagulação.
A trombina gerada durante o processo de coagulação aumenta a atividade coagulante. Várias clivagens adicionais geram peptídeos de 50, 43 e 73 kDa, produzindo uma atividade coagulante. O fator VIII é inativado quando o PCA atua sobre a arginina na posição 336, formando uma banda de 59kDa e uma banda de 43kDa. O fator Xa o inativa por proteólise em arginina 336 e 1721, gerando uma banda de 67 kDa. A trombina o inativa pela ativação da proteína C, que é seu inibidor fisiológico. Por si só, o fator VIII é uma molécula instável altamente reativa. É estabilizado no plasma pela ligação ao fator de von Willebrand.

VIA INTRÍSECA
A via intrísica começa quando o fator XIII entra em contato com a matriz subendotelial carregada negativamente, passando por uma mudança conformacional que dá origem à variedade ativa XIIa. A referida reação é acelerada na presença de cinogênio de alto peso molecular. O XIIa, por sua vez, ativa a pré-calicreína que é convertida em calicreína, uma enzima que converte um grande número de fator XII em XIIa, que por sua vez transforma o fator IX em IXa. Em uma etapa subsequente, IXa forma um agregado enzimático com fosfolipídios da membrana plaquetária, cálcio e fator VIIIa, para converter o fator X em Xa. Este último forma o complexo da protrombinase com os fosfolipídios fornecidos pelas plaquetas, cálcio e fator Va, responsáveis por transformar a protrombina em trombina, um dos mais poderosos ativadores da agregação plaquetária. Ao mesmo tempo, ele transforma os fatores VIII e V em suas variedades ativas VIIIa e Va. Da mesma forma, ele converte centenas de moléculas de fibrinogênio em fibrina, para formar o tampão hemostático. Finalmente, ativa o fator XIII para dar origem ao XIIIa, uma molécula que permite a formação de ligações cruzadas entre as moléculas de fibrina para formar uma estrutura polimétrica estável.
VIA EXTRINSECA
A via extrínseca começa com a interação do fator tecidual (uma proteína sintetizada por células endoteliais e macrófagos que migraram intimamente) com o fator VII para dar origem ao fator VIIa. Este, por sua vez, ativa diretamente o fator X para gerar Xa, com a consequente produção de trombina, que segue o caminho indicado acima.
ABREVIATURAS
ADP – disfosfato de adenosina
ATP – trisfofato de adenosina
DAG – diacilglicerol
IP3 – fosfato de inositol 3
MCL – cadeia leve de miosina
MCP – fosforilação de cadeia leve de miosina
MK – miosina quinase
PDGF – fator de crescimento derivado de plaquetas – fator de crescimento derivado de plaquetas
PKC – proteína quinase C
P-47 – kDa – peptídeo 47 kDa
PGG2 – prostaglandina G2
PGH2 – prostaglandina H2

whoah this weblog is excellent i love studying your articles.
Keep up the great work! You already know, lots of persons are searching round for this info, you can aid them
greatly.
Florian,Thank you very much for your comment.
My intention is precisely to help inform people about this topic of great promotion, our health.
Gratitude!
Em português:
Agradeço muito seu comentario.
Minha intensão é justamente ajudar a informar as pessoas com esse tema de muita promoção, nossa saúde.
Gratidão!