Por Raquel Luiza,

Quadro clínico caracterizado pela produção exagerada do hormônio do crescimento (GH é a sigla em inglês).
A acromegalia é uma doença crónica rara, causada pela hipersecreção de hormona do crescimento (GH) e do fator de crescimento semelhante à insulina tipo 1 (IGF‐1). O diagnóstico da doença o mais precocemente possível é essencial para prevenir as suas complicações e morbilidade. Apresentamos o caso de uma mulher, enviada à nossa consulta de endocrinologia por obesidade, com escassas características acromegaloides, mas com várias comorbilidades desta patologia. Apesar do tempo de evolução até ao diagnóstico, do tamanho do tumor e de se tratar de um tumor misto, a doente está, neste momento, curada.
A hormona do crescimento é secretada pelas células somatotrópicas da hipófise e é necessária para o crescimento do corpo humano. Além disso, atua no metabolismo estimulando a incorporação de aminoácidos às proteínas, aumentando a liberação de ácidos graxos e inibindo a formação de glicose pelos tecidos.
O excesso de GH produz um aumento exagerado no crescimento dos ossos e tecidos moles.
A causa mais comum são os adenomas hipofisários, tumores benignos da glândula pituitária, que podem invadir localmente e têm capacidade de secretar hormônios.
Se o excesso do hormônio ocorre em crianças, antes do fechamento das epífises ósseas, ou seja, antes do término do crescimento vertical, o quadro clínico é denominado de gigantismo e é caracterizado por crescimento excessivo de ossos, músculos e órgãos, e por altura estatura. São crianças extremamente grandes para a idade e têm início tardio da puberdade.
A acromegalia ocorre quando o excesso de hormônio do crescimento ocorre após o fechamento das epífises ósseas. Esta doença evolui lentamente.
Causas da acromegalia
Na grande maioria dos casos, a acromegalia é provocada pelo desenvolvimento de um adenoma hipofisário, tumor benigno na hipófise, glândula situada no cérebro que aumenta de tamanho e eleva sua produção do hormônio do crescimento. Em situações mais raras, a acromegalia pode ser provocada por hiperatividade da hipófise. A doença também pode ser hereditária ou, em outros casos, ter causa desconhecida.
Quais são os sintomas?
A hipersecreção de GH e IGF-1 induz um aumento das partes acrais característico desta doença, podendo ocorrer manchas cutâneas (formações polipoides). A face desses pacientes é caracterizado pela presença de prognatismo, má oclusão dentária, diastemas (separação dos dentes) e macroglossia;
O nariz é mais largo, os lábios são mais grossos e os arcos supraobitais são proeminentes,
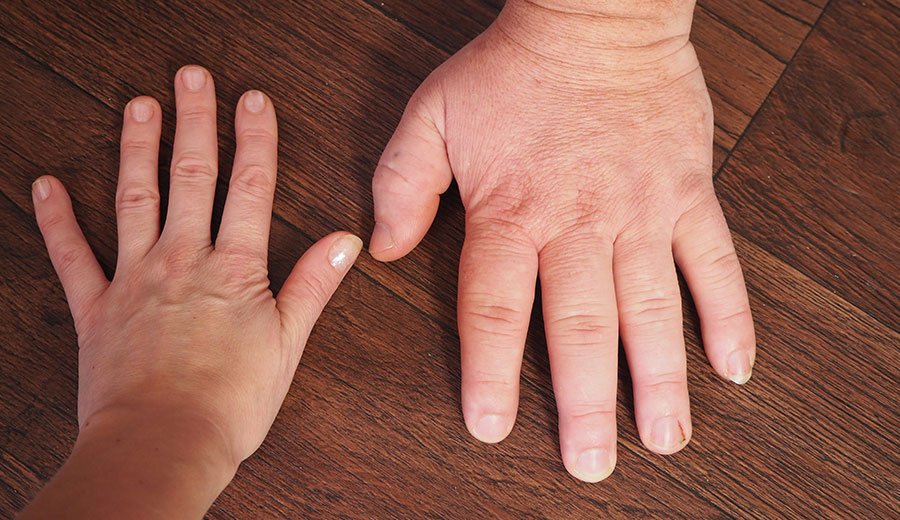
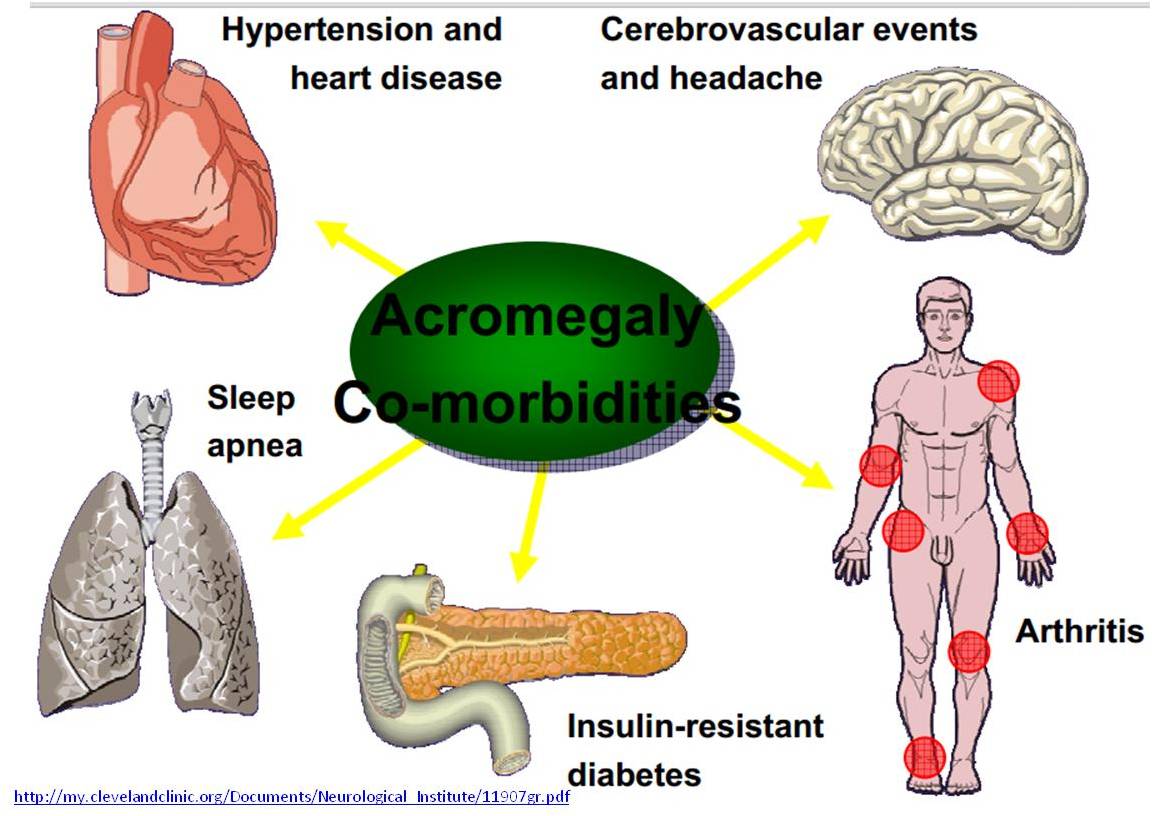
As mãos e os pés têm aparência desajeitada devido ao seu aumento e é comum os pacientes precisarem aumentar o tamanho dos sapatos e anéis. Pacientes com acromegalia geralmente apresentam hiperidrose e pele oleosa devido ao aumento da secreção de sebo. A voz é rouca devido ao espessamento da laringe e das cordas vocais. 50% dos pacientes apresentam apneia obstrutiva durante o sono, devido à macroglossia e espessamento da faringe e laringe.
A hipertrofia dos tecidos moles é comum para produzir neuropatias de encarceramento, sendo a mais comum a síndrome do túnel do carpo. As vísceras também hipertrofia e, em alguns pacientes, bócio difuso ou multinodular (que geralmente é eutireoidiano), hepatomegalia, esplenomegalia, nefromegalia e / ou hiperplasia prostática podem ser detectados.
Como é realizado o diagnóstico?
Os níveis de hormônio do crescimento no sangue estão elevados.
É útil determinar a somatomedina C (IGF-1) no sangue, uma molécula que estimula diretamente o crescimento induzido pelo GH.
O teste de supressão da secreção de GH após sobrecarga de glicose é anormal, visto que a diminuição fisiológica dos níveis de hormônio do crescimento não ocorre devido ao aumento da concentração de glicose no sangue.
A tomografia axial computadorizada e a ressonância magnética nuclear permitem avaliar a anatomia dos processos.
Uma avaliação completa da função dos outros hormônios hipofisários deve ser realizada.
Tratamento
O tratamento de escolha para a acromegalia do adenoma hipofisário é a cirurgia, que oferece a possibilidade de normalização imediata dos níveis de GH e cura definitiva da doença em um alto percentual de pacientes.
Em pacientes não curados ou nos quais o risco cirúrgico é muito alto, o tratamento de escolha são os análogos da somatostatina: octreotida ou lanreotida.
Os agonistas da dopamina foram os primeiros medicamentos usados no tratamento da acromegalia; a droga de escolha é a cabergolina. O tratamento combinado de agonistas da dopamina com análogos da somatostatina normaliza os níveis de GH e IGF-1 em alguns pacientes que não respondem ao último.
Os antagonistas do receptor de GH (pegvisomant) são indicados em pacientes que não respondem ou são intolerantes aos análogos da somatostatina e agonistas da dopamina.
A radioterapia tem sido o tratamento mais usado em pacientes que não são curados por cirurgia. Previne o crescimento do tumor na maioria dos pacientes e normaliza os níveis de GH em 75% dos casos, e a principal limitação é que essa eficácia leva vários anos para ser alcançada.