
Por Raquel Luiza,
O que é ?
Defeito genético que acarreta na falha de transpore hepatocelular do cobre, que não é excretado na bile e é acumulado no fígado e outros órgãos, incluindo Sistema Nervoso Central.
Causa
É uma doença genética autossômica recessiva, devido à mutação do gene ATP7B. Ou seja, a pessoa nasce com a doença. Esta mutação faz com que menos cobre seja eliminado com a bile, causando um excesso de cobre no fígado e no restante do corpo.

Quais são os fatore de risco?
É uma doença genética, ou seja, não é adquirida. A pessoa nasce com esta alteração.
Qual a Incidência?
A incidência é de 1 para cada 30.000 pessoas, e afeta populações em todo o mundo. Afeta igualmente homens e mulheres, mas mulheres são mais suscetíveis a desenvolver insuficiência hepática aguda, e homens de ter alterações psiquiátricas.
O diagnóstico costuma ser dado entre 5 e 35 anos.
Crianças costumam apresentar mais alterações hepáticas, enquanto adultos abrem o quadro clínico com sintomas neurológicos.
Quais são os sintomas e sinais que aparecem?
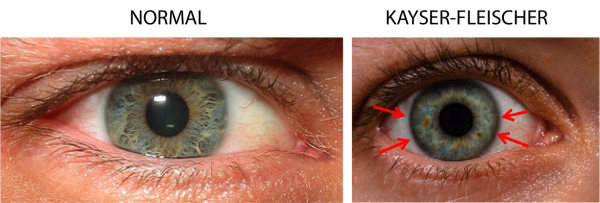
Amplo expectro de apresentação clínica. Os sintomas neurológicos masi comum são disartria, ataxia e distonia. Geralmente há elevação de transaminases e o paciente pode cursar com dor abdominal, insuficiência hepática aguda e cirrose hepática. Sintomas psiquiátricos como irritabilidade, psicose e depressão ocorrem. Os Pacientes apresentam os clássicos anéis de Kayser-Fleisher (foto abaixo) nos olhos.

Como é realizado o diagnóstico?
O diagnóstico é realizado através da história clínica e dos exames de sangue. Pacientes que possuem suspeita de Doença de Wilson, devem realizar exames de função hepática, hemograma, coagulograma, nível de ceruloplasmina e cobre no sangue, nível de cobre urinário e exame dos olhos na procura de anéis de Kayser-Fleischer.
Exames de Sangue: Elevação leve a moderada (> 2x) dos exames da função do fígado como o TGO e o TGP e baixos níveis de ceruloplasmina (< que 20mg/dL em 90% dos casos) são os achados mais encontrados. O TGO geralmente é um pouco mais elevado que o TGP. Sinais laboratoriais de cirrose, como plaquetopenia, elevação de bilirrubina, coagulopatia (RNI elevado), hipoalbuminemia também podem ser encontrados. Alguns pacientes podem apresentar exames que sugerem hemólise, com coombs negativo. Anemia com teste de coombs negativo pode ser evidenciado. Outros testes podem ser realizados, como: cobre urinário em 24 horas >100mcg, cobre no sangue baixo e o teste genético para avaliar a mutação do gene ATP7B.
Ecografia: Não ajuda no diagnóstico da doença de Wilson em si. Pode diagnosticar sinais de cirrose, como as alterações no parênquima do fígado, esplenomegalia e circulação colateral.
Tomografia e ou Ressonância: A tomografia e a ressonância abdominal podem demonstrar sinais de cirrose, porém não determina a causa da mesma. A ressonância cerebral, pode demonstrar alterações sugestivas de doença de Wilson.
Biópsia Hepática: A biópsia pode demonstrar acúmulo de cobre.
Tratamento
É realizado com D-penicilamina ( 250 – 500 mg/dia, aumentado para 1000- 1500 mg/dia gradativamente). O tratamento deve se monitorado, e a dose diminuída após controle laboratorial da doença, para doses de manutenção. Opção: Trientina, 20 mg/kg/dia. A manutenção da doença também pode ocorrer com sais de zinco. Durante o tratamento inicial, deve-se evitar dieta rica em cobre.
Prognóstico
O prognóstico em pacientes que fazem o tratamento é excelente, mesmo em pacientes com certo grau de comprometimento do fígado e neurológico. Os sintomas melhoram gradualmente com o tratamento. Sem tratamento, todo o paciente vai a óbito, principalmente de doença no fígado. Pacientes que desenvolvem falência hepática aguda falecem em até 95% dos casos, se não houver um transplante.
Como evitar?
Não há como evitar, pois é uma doença genética.
